The speed of reactions
Now we turn to the speed of chemical processes. Speed matters.
Consider, for example, the hydrolysis of Adenosine triphosphate (ATP) to Adenosine diphosphate, the reaction from which all living cells derive their energy to operate. In this reaction, a 3-phosphate chain attached to an adenine-sugar complex is trimmed by one phosphate (OPO3) group by hydrolysis to form the di-phosphate. The process liberates energy, which is used in cellular processes.
In particular, muscle movement requires a great deal of ATP to be stored in muscle cells and to be converted rapidly to ADP to release the energy needed for muscle contraction. You wouldn't want it to take forever for your muscles to fire when faced with a saber-tooth tiger. That reaction has to be FAST.
Well, if we mix ATP with water in a beaker, we'd have to wait an awfully long time for anything to happen. The reaction is VERY slow. Its reaction profile looks like this:
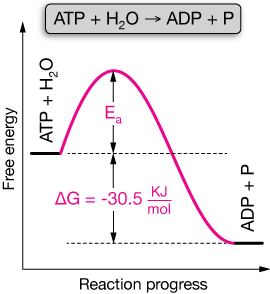
Notice that $\Delta G$, the change in the Gibbs energy, is negative, so this is a spontaneous reaction. That hardly matters, however, if the reaction is very slow. For all practical purposes, it doesn't seem spontaneous.
This is true because the energy of activation ($E_a$) is large. In fact, to get this reaction to go, cells use a set of enzymes (catalysts) called ATP-ases. These effectively lower the activation energy barrier to let the reaction proceed much more rapidly.
A rapid reaction
Now consider the combination of hydrogen gas and oxygen gas to make water,
$$2 \, H_2 + O_2 \longrightarrow 2 \, H_2O$$
This reaction is also thermodynamically favorable — exothermic and spontaneous. But take a look at this clip to see how fast it can go:
That's the Sept. 1, 2016 explosion of a SpaceX Dragon rocket on the launch pad. It was determined that a spark ignited the mixture of kerosene and oxygen inside the rocket (instead of at the nozzles, as intended). All of the fuel burned catastrophically in just a second or so. That's a fast reaction.
So we have a continuum of reaction rates, from very slow to very fast. Very often we'd like to speed up slow reactions and slow down fast reactions, and so we study what kinds of things affect reaction rates and how to model them mathematically.
This section will develop some of the main ideas in the field of chemical kinetics, and further sections will go into more detail.
Collision theory of reactions
Approximately-speaking, chemical reactions happen when reactants collide. Whether in the gas, liquid or solid phase, that's mostly how reactions occur.
Now it's not that simple, of course. First, there are exceptions, such as decomposition reactions where one compound falls apart – say when it's heated – no collision necessary. And second, it's not usually as simple as just a collision. Reacting compounds have to collide in the right orientation and with the right relative kinetic energy (not too slow, not too fast) to react. Take the reaction
$$Cl + HCl \rightleftharpoons ClH + Cl$$
for example. It's not a very interesting reaction; the products are the same as the reactants – just turned around, but it can help us illustrate this idea. Let's take a look at two kinds of collisions. In the first, The free chlorine (Cl) atom (blue) comes in the wrong way, and no reaction occurs:
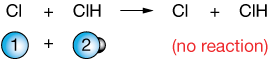
But when the Cl approaches the other "side" of HCl, the orientation is correct and the reaction occurs:
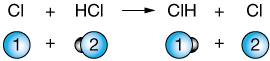
Still, simple collision theory is pretty helpful in understanding a lot of the features of reaction rates.
At high concentration, and assuming every collision leads to a reaction, we'd have a lot of reactions (yellow "flashes") per unit volume, because we'd expect many more collisions.
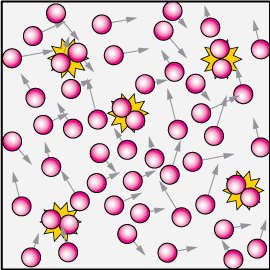
To think about that, imagine two blindfolded skaters on a rink. It's unlikely that they'll run into each other. Sure, it will happen once in a while, but not often.
Now put 100 blindfolded skaters out there and you'll have a lot more collisions. It's the same with collision theory and reactions.
If the concentration is reduced, there will be fewer reactions in an identical time.
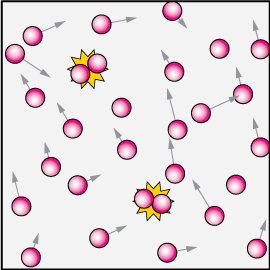
Now we can start thinking about reaction rates. Imagine the model reaction
$$A + B \longrightarrow AB$$
and imagine that half of the pink balls in our figures above are $A$s and half are $B$s. At high concentrations, the rate of collisions between $A$ and $B$ will be high, thus we'd expect the rate of reaction to be relatively high (even if just a small fraction of collisions actually led to a reaction). But as the concentrations of $A$ and $B$ decreases due to the formation of $AB$, we'd expect the rate of $A-B$ collisions to diminish, and thus for the rate to decrease. In the extreme, when there are only a couple of $A$'s and a couple of $B$'s, it might take them a while to find one-another and collide.
If we made a graph of reaction rate versus time, it might look like this:
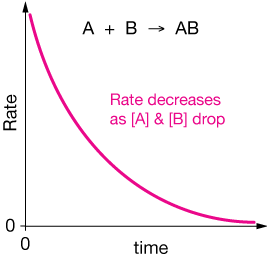
OK, that should give us a feel for why the rates of reactions change over time. Lower concentrations of reactants, as more reactants are turned into product(s), leads to fewer productive collisions in a given time, thus the rate is lower.
Modeling reaction rates
We need a mathematical model for reaction rates. We'll begin with the definition of speed, distance divided by time:
$$s = \frac{d}{t}$$
Now let's translate that to a simple reaction, reactant $A$ turning to product $B$:
$$A \longrightarrow B$$
Now we can express the "speed" or rate of this reaction in terms of how much reactant gets used up:
$$\text{rate} = \frac{\Delta[A]}{\Delta t} = \frac{[A]_f-[A]_i}{\Delta t}$$
Now the problem is that we can have all kinds of reactions, some like $A \longrightarrow B$, but also some in which two or more reactants come together to make products.
To account for such complications, we'll write a "rate law" that looks like this:
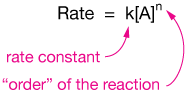
The rate constant, $k$ will be the proportionality constant (and, like equilibrium constants, will have situation-dependent units), and the power of the reactant concentration, $n$, we'll call the reaction "order."
When $n=0$, the reaction is zeroth order
when $n=1$, the reaction is first order; $n=2$ is second-order, and so on.
Rate laws can take a wide variety of forms. Here are some examples:
Some examples of rate laws
| Reaction | Rate law | Notes |
|---|---|---|
| $2 \, N_2O_{5 \, (g)} \rightarrow 4 \, NO_{2 \, (g)} + O_{2 \, (g)}$ | $\text{Rate } = k[N_2O_5]$ | The coefficient of N2O5 doesn't matter. |
| $2O_{3 \, (g)} \rightarrow 3 \, O_{2 \, (g)}$ | $\text{Rate } = k\frac{[O_3]^2}{[O_2]}$ | Rate laws can include concentrations of products. |
| $(CH_3)_3CBr + H_2O \rightarrow (CH_3)_3COH + HBr$ | $\text{Rate } = k[(CH_3)_3CBr]$ | Not all reactants have to appear in the rate law. |
| $2NH_{3 \, (g)} \rightarrow N_{2 \, (g)} + 3 \, H_{2 \, (g)}$ | $\text{Rate }= k$ | The rate is constant (zero order) |
Pro tip:
You may have the (correct) impression that there's no rhyme or reason to how rate laws as written. That's because they depend on the often-hidden details of the reaction.
Rate laws cannot be determined by looking at an overall reaction. They must be determined experimentally.
Zeroth-order reactions
When the rate law of a reaction depends on the 0th power of the concentration of a reactant, we say it's a zerothe-order (or zero-order) reaction, or simply that it is zero-order in one of the reactants.
Consider the rate law
$$ \begin{align} \text{rate} &= k \, [A]^0 \\[5pt] \text{rate} &= \text{constant} \end{align}$$
because $[A]^0=1$. The decomposition of ammonia gas,
$$2 \, NH_{3 \; (g)} \longrightarrow N_{2 \; (g)} + 3H_{2 \; (g)}$$
is a zero-order process. It doesn't depend on the concentration of ammonia. At some rate (depending on temperature), ammonia molecules just fall apart into $N_2$ and $H_2$.
If we plot the rate of a zero-order reaction vs. the concentration of reactant, it will look like this:
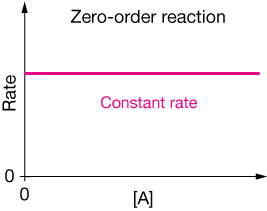
A reaction such as
$$A + B \longrightarrow AB$$
may be zero-order on only one of the reactants. For example, if the rate law for that reaction was
$$\text{rate} = [A][B]^0$$
we'd say that the reaction was "zero order in $B$," i.e. doesn't depend on the concentration of $B$, and "first-order in $A$.
First-order reactions
Consider a simple reaction like
$$A \longrightarrow B$$
The reaction is first-order if its rate law looks like
$$ \begin{align} \text{rate} &= k[A]^1 \\[5pt] \text{rate} &= \text{linear function of }[A] \end{align}$$
A plot of the reaction rate vs. the concentration of reactant A would look like this:
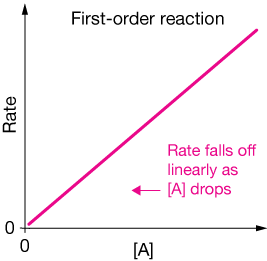
An example of a first order reaction is the conversion of methyl isonitrile to acetonitrile:
![]()
The rate law is
$$\text{rate} = k[CH_3 NC]$$
It's worth noticing a couple of things about this rate law.
- First, the rate needs to have units of moles/s, so the units of the rate constant k are 1/s or s-1, "per second."
-
Second, you might have noticed that in the previous section on zero-order reactions, the decomposition of ammonia did not depend on the concentration of ammonia. So why does the rearrangement of methyl isonitrile (CH3NC) depend on its concentration?
It turns out that the detailed mechanism for this interconversion (isomerization) depends, roughly-speaking, upon the interaction of one CH3NC with another. In that case, it's easy to see that as the concentration of CH3NC diminishes, so will the rate of the reaction.
2nd-order reactions
The general form of a rate expression for a reaction like
$$2 \, A \longrightarrow B$$
is
$$ \begin{align} \text{rate} &= k[A]^2 \\[5pt] \text{rate} &= \text{quadratic fctn. of }[A] \end{align}$$
The rate now depends upon the square of the concentration of the reactant (i.e. it depends quadratically). Here's a graph of the rate vs the concentration of $A$. Notice that as $[A]$ diminishes, so does the rate.
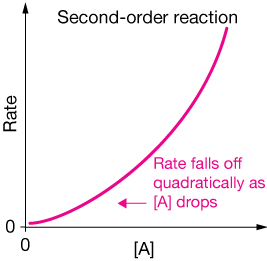
One example of such a reaction is the decomposition of $NO_2$ to NO and $O_2$ in the gas phase. This reaction requires an aligned collision of two $NO_2$ molecules in order to produce the products.
$$2 \, NO_{2 \; (g)} \longrightarrow NO_{(g)} + O_{2 \; (g)}$$
In that case, it's not difficult to imagine why the rate would depend on the square of the concentration:
$$\text{rate} = k[NO_2]^2$$
Again, the rate expression can't be determined from just looking at the chemical equation. It must be determined experimentally.
The rate of a reaction like
$$A + B \longrightarrow C$$
can also be second order: It can be first order in $A$ and in $B$, requiring a collision between $A$ and $B$ for the reaction to occur. The rate expression for such a reaction might look like
$$\text{rate} = k[A][B]$$
In this case, the reaction is said to be "first order in $A$ and $B$, but second order overall."
Examples
Now let's take a look at some experimental data, generally concentrations of one or more reaction components monitored over time, to see if we can determine some rate laws.
Example 1
Consider the gas-phase reaction between oxygen and nitric oxide:
$$O_{2 \; (g)} + 2 NO_{2 \; (g)} \longrightarrow 2 \, NO_{2 \; (g)}$$
Now suppose we've collected the following data. We have starting concentrations of O2 and NO, and we've measured "initial rates," rates of formation of product measured as close to the initiation of the reaction as possible:
| Starting | |||
|---|---|---|---|
| concentration | Rate | ||
| Expt. | [O2] | [NO] | mol·L-1·s-1 |
| 1 | $0.011$ | $0.013$ | $3.19 \times 10^{-3}$ |
| 2 | $0.021$ | $0.014$ | $5.81 \times 10^{-3}$ |
| 3 | $0.011$ | $0.029$ | $1.70 \times 10^{-3}$ |
The general form of the rate constant expression will be
$$\text{rate} = k[O_2]^m [NO]^n$$
Our job will be to determine $m, \; n$ and $k$ from the data. There are a couple of ways we can do this.
Method 1
In the first method, we'll notice that the data have been collected rather intelligently. In experiments 1 and 3, [O2] was held constant while [NO] was varied, and the opposite is true for experiments 1 and 2, at least to within some acceptable error in the NO concentrations.
We can make use of those facts. If we look at experiments 1 and 2 (hereafter E1 and E2), they represent a look at what happens when only [O2] is varied. The change in [O2] from E1 to E2 is 0.001 → 0.021, or a factor of 1.91. The change in initial reaction rate, 3.19 × 10-3 → 5.81 × 10-3 is roughly the same, a factor of 1.8. It's not exact, but it really looks like this reaction is first order in [O2].

Now let's do the same thing with E1 and E3, in which [O2] remains constant and [NO] is varied. Upon increasing [NO] by a factor of 2.23, the rate increased by a factor of 5.32.

Now a little tinkering tells us that the square root of 5.32 is 2.31. It looks like the rate of the reaction increases with the square of [NO]. So from this data, it looks like our rate equation is:
$$\text{rate} = k[O_2][NO]^2$$
The only thing that's left is to calculate the rate constant, $k$. We have all of the information we need to do that by rearranging the rate equation.
$$k = \frac{\text{rate}}{[O_2][NO]^2}$$
Using the data from E1, we get
$$ \begin{align} k &= \frac{3.19 \times 10^{-3}}{[0.011][0.013]^2} \\[5pt] &= 1716 \; \frac{L^2}{\text{mol}^2 \, s} \end{align}$$
Now a better way to evaluate $k$ might be to calculate it using all three sets of data and average the results, but we'll let that go for now.
Method 2
We can also solve for $m$ and $n$ algebraically. If we plug each of our three datasets into the general rate equation, we get:
$$ \begin{align} \color{#E90F89}{A} \; \; 3.19 \times 10^{-3} &= k[0.011]^m[0.013]^n \\[5pt] \color{#E90F89}{B} \; \; 5.81 \times 10^{-3} &= k[0.021]^m[0.014]^n \\[5pt] \color{#E90F89}{C} \; \; 1.70 \times 10^{-3} &= k[0.011]^m[0.029]^n \end{align}$$
Now dividing equation A into equation B gives us
$$ \require{cancel} \color{#E90F89}{\frac{B}{A}} = \frac{\cancel{k}[0.021]^m[0.014]^n}{\cancel{k}[0.011]^m[0.013]^n} = \frac{5.81 \times 10^{-3}}{3.19 \times 10^{-3}}$$
The resulting expression can be solved by taking a log of both sides and doing a little bit of algebra:
$$ \begin{align} 1.91^m &= 1.82 \\[5pt] m \cdot ln(1.91) &= ln(1.82) \\[5pt] m &= 0.93 \end{align}$$
That's pretty close to one. We can solve for $n$ in the same way, by dividing equation C by equation A:
$$\color{#E90F89}{\frac{C}{A}} = \frac{\cancel{k}\cancel{[0.011]^m}[0.014]^n}{\cancel{k}\cancel{[0.011]^m}[0.013]^n} = \frac{1.70 \times 10^{-3}}{3.19 \times 10^{-3}}$$
The same log and algebraic tricks yield $n$:
$$ \begin{align} 2.23^n &= 5.33 \\[5pt] n \cdot ln(2.23) &= ln(5.33) \\[5pt] n &= 2.09 \end{align}$$
Finally, rounding to $m = 1$ and $n = 2$, we can solve for $k$ in the same way, and we're done.
These methods are typical of evaluating kinetics data recorded in the lab. It's just not possible (though sometimes you might get lucky) to figure out these exponents, and thus the rate constant, $k$, without looking at experimental data like this. Which method you use is your choice.
Example 2
The following data were measured for the gas-phase reaction between nitric oxide (NO) and bromine (Br2),
$$2 \, NO_{(g)} + Br_{2 \; (g)} \longrightarrow 2 \, NOBr_{(g)}$$
| Expt. | [NO] | [Br2] | Rate* |
|---|---|---|---|
| $1$ | $0.10$ | $0.10$ | $12$ |
| $2$ | $0.10$ | $0.20$ | $23$ |
| $3$ | $0.20$ | $0.10$ | $49$ |
| $4$ | $0.30$ | $0.10$ | $110$ |
*
Our rate law will look something like
$$\text{rate} = k[Br_2]^m[NO]^n$$
and out job is to find out what $m$, $n$ and $k$ are. Notice that in the data table, [Br2] was held constant in experiments 1, 3 & 4 (E1, E3 & E4). One strategy is to make a plot of rate vs. the concentration that is not held constant. Here is a plot of rate vs. [NO]:
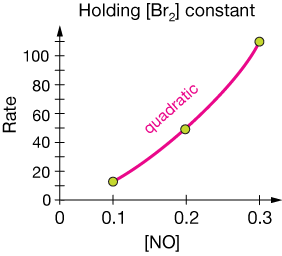
There is predictability there. Notice that when we double the concentration of NO, say from 0.1 to 0.2 the initial rate of the reaction increases four-fold (from 12 to 49). There's a quadratic relationship lurking in there, so we now know that the rate law will look something like
$$\text{rate}=k[Br_2]^m[NO]^2$$
Next we notice that there are two data points for which [NO] = 0.1, and that doubling [Br2] in that condition doubles the rate. The exponent is one. So our rate law has the form
$$\text{rate}=k[Br_2][NO]^2$$
Now we just have to find k by rearranging that rate expression:
$$k = \frac{\text{rate}}{[Br_2][NO]^2}$$
We can do that for each of the data points to obtain four values of $k$:
- $k = 12000$
- $k = 11500$
- $k = 12250$
- $k = 12222$
The fact that all of these are so close (the average is 12000) means that we've correctly determined the exponents.
The reaction is first-order in Br2, second-order in NO and third-order overall. The units of the rate constant are L2·mol-2·s-1.
Practice problems
Using the data provided for the reaction, find the rate law and the rate constant, $k$, for each of the following reactions:
$1. \; \; 2 \, NO_{(g)} + Cl_{2 \; (g)} \longrightarrow 2 \, NOCl_{(g)}$
| Expt. | [NO] (M) | [Cl2 (M) | Rate (M/s) |
|---|---|---|---|
| 1 | 0.0290 | 0.0110 | 3.4 × 10-4 |
| 2 | 0.0145 | 0.0100 | 8.5 × 10-5 |
| 3 | 0.0148 | 0.0390 | 3.4 × 10-4 |
Solution
Look for data that holds one concentration constant and use that to determine the effect of the concentration that changes. The Cl2 concentration is the same in the first two rows.:

If we double [NO] the rate increases by a factor of 4, or 22. So the exponent of [NO] is 2.
Now in the bottom two rows, [NO] is relatively constant:

Notice that when [Cl2] is increased by a factor of 4, the rate increases by the same factor, so the exponent of [Cl2] is 1. And the rate law is
$$\text{rate} = k[NO]^2[Cl_2]$$
We can find $k$ by using any of the three rows of data:
$$ \begin{align} 3.4 \times 10^{-4} &= k[0.0290]^2[0.0110] \\[5pt] k &= \frac{4.3 \times 10^{-4}}{[0.0290]^2[0.0110]} \\[5pt] &= 36.8 \end{align}$$
So the rate law is
$$\text{rate} = 36.8[NO]^2[Cl_2]$$
We could also solve this algebraically using the rows in the data table:
$$ \begin{align} \color{#E90F89}{A}: \; k[0.0290]^m[0.0110]^n = 3.4 \times 10^{-4} \\[5pt] \color{#E90F89}{B}: \; k[0.0145]^m[0.0100]^n = 8.5 \times 10^{-5} \\[5pt] \color{#E90F89}{C}: \; k[0.0148]^m[0.0390]^n = 3.4 \times 10^{-4} \end{align}$$
Now the ratio of lines A to B is
$$\color{#E90F89}{A/B}: \frac{\cancel{k}[0.0290]^m[0.0110]^n}{\cancel{k}[0.0145]^m[0.0100]^n}$$
The $k$s cancel to give
$$ \begin{align} \left[ \frac{0.0290}{0.0145} \right]^m \left[ \frac{0.0110}{0.0100} \right] &= 4 \\[5pt] 2^m \cdot 1.1^n &= 4 \end{align}$$
Using the laws of logs and approximating $1.1 \approx 1$, we get
$$ \begin{align} ln(2^m \cdot 1^n) &= ln(4) \\[5pt] m \cdot ln(2) + n \cdot ln(1) &= ln(4) \\[5pt] m = \frac{ln(4)}{ln(2)} = 2 \end{align}$$
Doing the same thing with the ratio of line C to line B gives us the other exponent:
$$ \begin{align} \color{#E90F89}{\frac{C}{B}}: \frac{\cancel{k}[0.0148]^m[00390]^n}{\cancel{k}[0.0145]^m[0.0100]^n} &= \frac{3.4 \times 10^{-4}}{8.5 \times 10^{-5}} \\[5pt] \left[ \frac{0.0148}{0.0145} \right]^m \left[ \frac{0.0390}{0.0100} \right]^n &= 40 \\[5pt] 1^m \cdot 3.9^n &= 4 \\[5pt] ln(1^m \cdot 3.9^n) &= ln(4) \\[5pt] m \cdot ln(1) + n \cdot ln(3.9) &= ln(4) \\[5pt] n &= \frac{ln(4)}{ln(3.9)} \approx 1 \end{align}$$
which gives us the same rate law as before.
$2. \; \; H_{2 \; (g)} + 2 \, ICl_{(g)} \longrightarrow I_{2 \; (g)} + 2 \, HCl_{(g)}$
| Expt. | PH2 (torr) | PICl (torr) | Rate (M/s) |
|---|---|---|---|
| 1 | 248 | 325 | 1.34 |
| 2 | 250 | 83 | 0.332 |
| 3 | 50 | 328 | 0.269 |
Solution
Look for data that holds one concentration constant, and use that to determine the effect of the concentration that does change. The Cl2 conc. is the same in the first two rows:

Notice that if we quadruple the partial pressure of ICl, the rate quadruples. Thus we know that the rate law is proportional to $[ICl]^1$. Now in the bottom two rows, [NO] is constant:

Notice that when [H2] is increased by a factor of 4, the rate increases by the same factor, so the exponent of [H2] is one, and the rate law is:
$$\text{rate}=k [ICl][H_2]$$
We can find $k$ by using an of the rows in the data table:
$$ \begin{align} 1.34 &= k[325][248] \\[5pt] k &= \frac{1.34}{[325][148]} = 2.78 \times 10^{-5} \end{align}$$
So the rate law is
$$\text{rate} = 2.78 \times 10^{-5}[ICl][H_2]$$
Or in terms of partial pressures we'd write
$$\text{rate} = 2.78 \times 10^{-5}P_{ICl} P_{H_2}$$
$3. \; \; 2 \, N_2O_{5 \; (g)} \longrightarrow 4 \, NO_{2 \; (g)} + O_{2 (g)}$
| Expt. | [N2O5] (M) | Rate (M/s) |
|---|---|---|
| 1 | 1.28 × 102 | 22.5 |
| 2 | 2.56 × 102 | 45.0 |
Solution
Notice from the data table that if we double [N2O5] the rate also doubles:

So the rate law will have the form
$$\text{rate}=k[N_2 O_5]$$
Now we can find $k$ from either line of data; we'll use the first:
$$ \begin{align} k[128]&=22.5 \\[5pt] k &= \frac{22.5}{128} = 0.176 \end{align}$$
So the rate law is
$$\text{rate}=0.176[N_2O_5]$$
$4. \; \; H_{2 \; (g)} + 2 \, ICl_{(g)} \longrightarrow I_{2 \; (g)} + 2 \, HCl_{(g)}$
| Expt. | PH2 (torr) | PICl (torr) | Rate (M/s) |
|---|---|---|---|
| 1 | 248 | 325 | 1.34 |
| 2 | 250 | 83 | 0.332 |
| 3 | 50 | 328 | 0.269 |
Solution
Look at the table of data and notice that when PICl is quadrupled, the rate quadruples, too.

So the exponent of the partial pressure of ICl is one. Now when the partial pressure of H2 is multiplied by 5, so is the rate, so that exponent is also one.
Our rate law has the form
$$\text{rate}=k \, P_{H_2} P_{ICl}$$
We can find $k$ from any of the rows of data. Using the first gives
$$ \begin{align} k(248)(325) &= 1.34 \\[5pt] k &= \frac{1.34}{248\cdot 325} = 1.662 \times 10^{-5} \end{align}$$
So the rate law is
$$\text{rate}=1.662 \times 10^{-5} P_{H_2} P_{ICl}$$
Problem 1 solution


![]()
xaktly.com by Dr. Jeff Cruzan is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. © 2012-2025, Jeff Cruzan. All text and images on this website not specifically attributed to another source were created by me and I reserve all rights as to their use. Any opinions expressed on this website are entirely mine, and do not necessarily reflect the views of any of my employers. Please feel free to send any questions or comments to jeff.cruzan@verizon.net.